| A New DNA Sequence Search - Compressive Genomics |
| Written by Alex Armstrong |
| Wednesday, 25 July 2012 |
|
With the improvements in sequencing technology we are rapidly approaching bioinformatic overload. Biologists need new methods to store and access the huge wave of data that is about to hit the subject and one solution might be to use compression At the moment DNA sequencing is following a doubling law with the speed doubling every few months. This far outstrips the rate at which computing power is increasing. The amount of genomic data is estimated to be increasing ten-fold each year and currently sequencing a complete human genome takes about a week and costs less than $10,000. Soon complete sequencing will be a matter of routine.
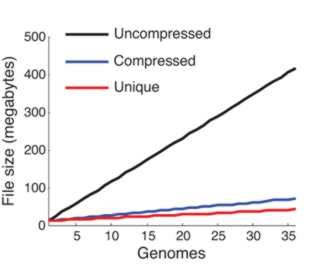
We clearly need some new ways of processing this data mountain. The main problem is searching through long sequences to find similar target sequences. You might think that searching one string for a target string is an easy algorithm, but the matching criterion is not exact. There can be mismatched point mutations and missing or additional units, indels (insertions/deletions). This makes matching sequences very difficult because you aren't searching for an exact match but for a good match. What makes the whole situation worse is the size of the search space and the target. We do know of exact solutions to the problem based on dynamic programming but in most cases practical analysis depends on heuristics. Put simply genome sequence analysis is a difficult algorithmic problem. The key to finding a solution is to notice that most genomic sequences differ by very little. It may well be that the number of complete genome sequences being stored is increasing rapidly, but the actual amount of new data is very small. In other words, a single DNA sequence isn't particularly compressible but a set of sequences shares so much in common that the redundancy can be used to store them in a much smaller storage space.
From the compression achieved you can see that adding more genomes adds very little information
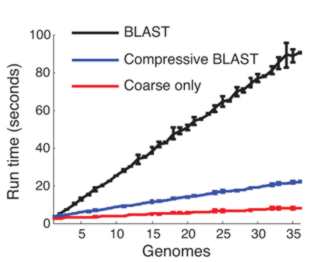
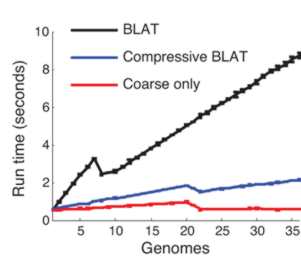
This compressed data format not only saves storage space it can make sequence analysis much faster. The trick is to use a compression format that respects the underlying structure of the situation by storing the details of substitution, deletion, insertion and so on. Once you have a compressed format sequence it can be searched in a time that is proportional to the file size without having to decompress it. In a recent paper in Nature Biotechnology, Po-Ru Loh, Michael Baym and Bonnie Berger (MIT and Harvard Medical School) have demonstrated that this can be made to work. A genomic database was compressed using sequence similarities and then the standard BLAST and BLAT algorithms were implemented on the compressed data without performing decompression. BLAST, for example, works by searching for small marker "words" taken from the target in the search sequence. If the words are found and they have the same spacing in both the target and the search sequence then we have a potential match. The compressed version of the algorithm first searches any parts of the search sequence that hasn't been compressed using a low threshold for a match. Then regions that have been identified as possible areas are effectively decompressed and searched with a higher threshold. The resulting algorithm was four times faster than the standard algorithm.
There are problems with the method. In particular as more sequences are added to the database the compression becomes less effective as the redundancy decreases. There is clearly a lot of work still to be done. In particular there is a need to study the compression methods that could be used and how they interact with the search. It is obvious that precomputing similarities between sequences as part of a dictionary style compression should make searching faster.
More InformationRelated ArticlesNYT on the Future of Computing Pancake flipping is hard - NP hard
Comments
or email your comment to: comments@i-programmer.info
To be informed about new articles on I Programmer, install the I Programmer Toolbar, subscribe to the RSS feed, follow us on, Twitter, Facebook, Google+ or Linkedin, or sign up for our weekly newsletter.

|
| Last Updated ( Wednesday, 25 July 2012 ) |